


Sommaire
- Frontière Part I/ Part II des BPF : Modalités d’application aux produits biologiques
- Moving One Unit Operation At a Time Toward Continuous Biomanufacturing
- « Close Collaboration Maximizes Value of Engineered Solutions and Saves Time in Start-Up »
- Improving Single Use Bioreactor Design and Process Development. New Research Towards Intensifying Seed- Train & Scale-up Methods Using 5:1 Turn- Down
- Qualification approach for the validation of real-word shipping in single-use systems
- Expansion of Human Bone Marrow-Derived Mesenchymal Stem Cells in BioBLU 0.3c Single-Use Bioreactors
- Single Use & Stainless Steel: complementarity or fight?
- Low Endotoxin Recovery (LER) is today one of authorities serious concerns regarding pyrogen testing
Suite aux questionnements des Entreprises Françaises de Biotechnologie sur la segmentation proposée par les inspecteurs des parties I et II des BPF vis-à-vis du procédé de production des médicaments biologiques, cet article a pour objectif de proposer une clarification argumentée du cas particulier de ces substances actives biologiques dont la formulation est réalisée lors de la production de substance active et non au moment de la fabrication du produit fini.

Ce principe s’applique pleinement aux protéines recombinantes et anticorps monoclonaux, et au cas par cas des procédés de fabrication, pour d’autres types de médicaments de biotechnologie et thérapeutiques innovantes (vecteurs de thérapie génique et vaccins, …).
Problématique
Le contexte
Lors de la production d’un composé chimique, les étapes de synthèse de la substance active (DS) et de mise sous forme pharmaceutique du produit fini (DP) sont séparées dans le temps et souvent géographiquement. Lors de la production d’une substance biologique, et plus particulièrement pendant sa purification, il est indispensable dans la majorité des cas d’ajouter instantanément des agents permettant sa stabilisation. En effet, une substance biologique n’étant pas stable, elle risquerait de se dénaturer et ainsi de perdre sa conformation native ou de subir très rapidement des dégradations. La dégradation de la protéine d’intérêt ou sa dénaturation conduirait à une perte de son efficacité thérapeutique ou à un risque d’immunogénicité.
La problématique discutée dans ce document consiste par conséquent à proposer une clarification des limites d’application des deux parties du référentiel réglementairement opposable pour :
• la fabrication et le conditionnement sous forme vrac de la substance active biologique, d’une part,
• la fabrication du médicament biologique à partir du vrac de la substance active jusqu’à son conditionnement final, d’autre part.
De ce fait, les modalités d’application de ce référentiel ont des conséquences sur le statut réglementaire en France du site fabricant.
La chaîne de fabrication d’un produit biologique
La particularité d’une chaîne de fabrication de produit biologique réside dans la nécessité de réaliser une formulation de la substance active au cours des étapes de purification ou d’obtenir un vrac stabilisé pouvant être stocké.
Ainsi, c’est la plupart du temps une substance active formulée qui est libérée par le site de production de la substance active.
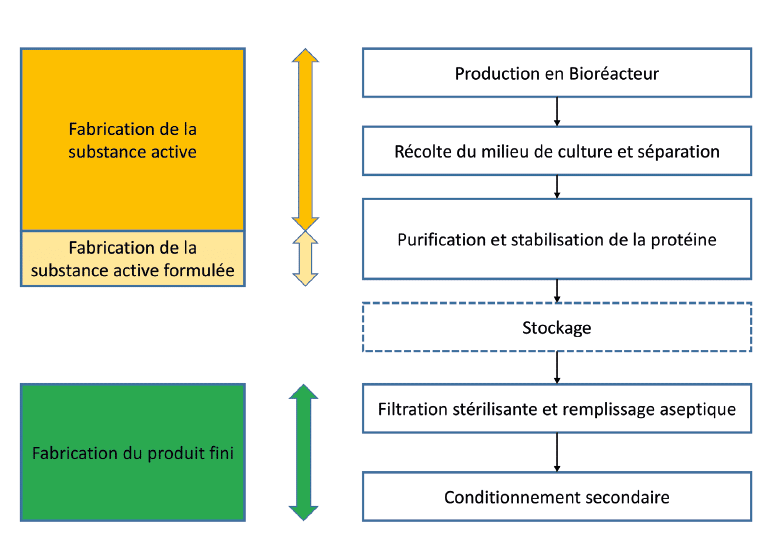
La formulation des substances actives biologiques
La formulation d’une substance active biologique, telle qu’un anticorps monoclonal par exemple, consiste principalement en sa stabilisation.
La substance biologique étant vulnérable aux facteurs extérieurs, l’étape de formulation est donc réalisée dès sa purification, par l’inclusion des stabilisants dans les tampons de purification.
L’ajout des composants en fin de purification est nécessaire afin de stabiliser l’anticorps ou la protéine thérapeutique peu de temps après son extraction.
Il s’agit également de réduire au maximum les étapes de congélation/décongélation et de stockage du produit, afin de diminuer le risque de dénaturation du produit et de limiter les stades de “Produit Intermédiaire”.
De ce fait, il est fréquent que cette stabilisation en cours de procédé soit la dernière étape de fabrication du produit avant la filtration stérilisante et la répartition du médicament.
Le produit qui est alors libéré par le fabricant est une substance active formulée, et non pas une substance active pure, comme dans le cas des produits chimiques. Cet état des lieux pose la question du niveau de segmentation entre la Partie I et la Partie II des BPF.
Discussion
Partie I et Partie II des BPF
Dans ce contexte des produits biologiques, il est nécessaire de repréciser le champ d’application de chaque partie de ce référentiel, sachant que l’annexe 2 des BPF couvre l’ensemble du processus de fabrication de ces médicaments biologiques et le niveau d’exigence BPF associé.
En France, la réglementation stipule clairement que l’application de la Partie I implique la notion d’établissement pharmaceutique fabricant.
L’application des BPF Partie II à des stades plus ou moins précoces de production de la substance active n’implique pas d’être établissement pharmaceutique.
La recommandation du groupe Bioproduction du Leem est résumée sur le schéma suivant qui compare les étapes de fabrication d’un médicament biologique à celles d’un médicament chimique et propose le niveau de segmentation entre les deux parties des BPF pour les deux types de produits.
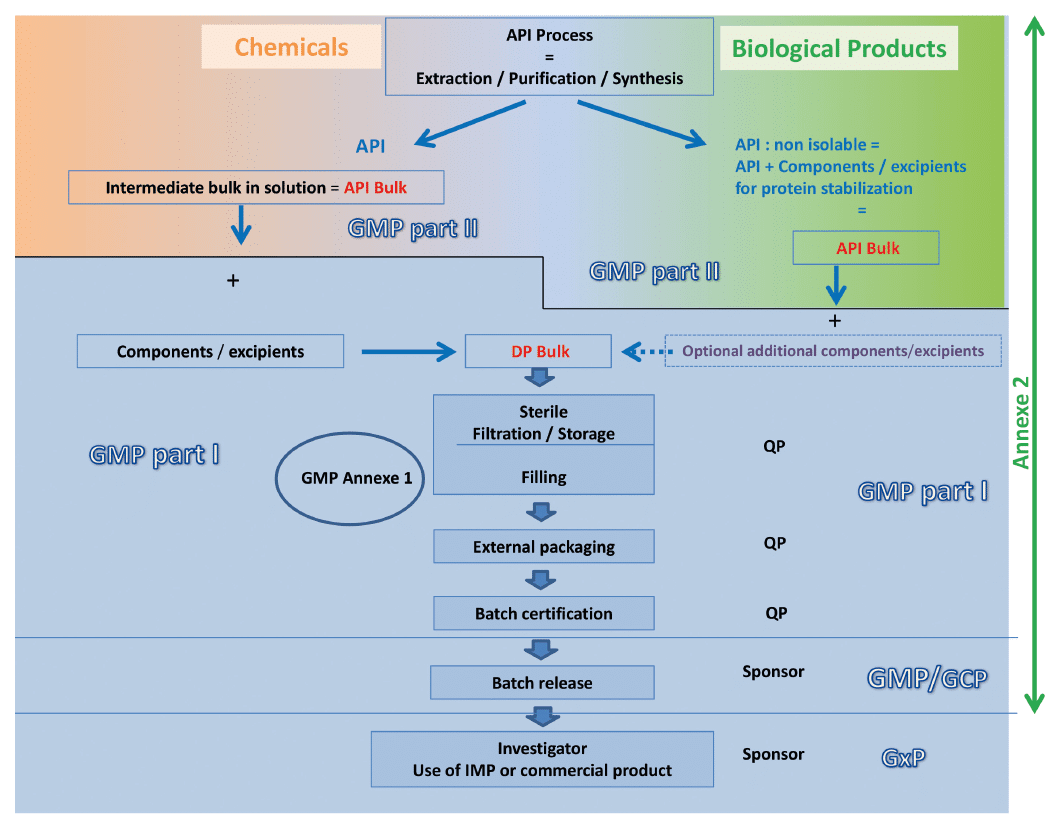
Notre lecture des BPF est basée sur les points suivants :
• Les excipients sont nécessaires pour la stabilisation de la protéine.
• Les excipients sont généralement indissociables du procédé de fabrication de la substance active.
• Le statut de la substance active conditionnée en vrac est revendiqué “low bioburden” plutôt que “stérile” (même si une filtration 0.2μ est effectuée) car la filtration stérilisante est réalisée par la suite, dans les étapes de fabrication du produit fini.
• Il est également possible de faire la différence entre la substance active formulée et la fabrication du PF par la présence d’une étape de stockage dans le procédé, les étapes de stockage donnant lieu à des tests de stabilité et de validation du stockage stérile.
• La fabrication de la substance active jusqu’à son conditionnement est décrite dans la partie”Substance” du dossier réglementaire (soumis aux BPF Partie II) alors que la fabrication du médicament (Produit Fini / Drug Product) est décrite dans la partie “Produit” (soumis aux BPF Partie I). Ceci permet ainsi d’assurer la cohérence entre le dossier d’AMM et l’inspection.
Ainsi, comme mentionné dans l’annexe 2, l’application de la Partie I des BPF concerne le processus de fabrication du produit fini, à partir de la filtration stérilisante ou d’une étape de stockage, c’est-à-dire le remplissage aseptique dans sa totalité (filtration + répartition + intégrité des unités à injecter). Lorsque le procédé de fabrication d’un produit biologique ne comporte pas d’étape de stérilisation, la Partie I des BPF s’applique à des stades amont définis au cas par cas.
Le fabricant de substance active formulée dans le cas des produits biologiques, n’est donc pas dans l’obligation d’avoir le statut d’établissement pharmaceutique.
Remarque sur les contrôles de l’entité biologique : compte-tenu de la spécificité des étapes de fabrication d’un produit biologique, la substance active se trouve être généralement de même composition que le produit fini. Ceci peut justifier que les contrôles analytiques spécifiques du produit fini, nécessitant des équipements particuliers et une expertise, puissent être faits sur le site de fabrication de la substance active sans que celui-ci ne soit établissement pharmaceutique mais en tant que sous-traitant de l’établissement pharmaceutique libérant le médicament.
Conclusion
La règlementation française exige que les fabricants de principe actif pharmaceutique répondent à la demande d’autorisation d’activité de fabrication de substance active auprès de l’ANSM pour la production de produit vrac, sans que ceux-ci ne prennent le statut d’établissement pharmaceutique.
Autrement dit, il n’est pas nécessaire d’être établissement pharmaceutique pour fabriquer une substance active biologique formulée et les entreprises du médicament confirment qu’il est cohérent de ne revendiquer l’application de la Partie 1 des BPF qu’à partir de l’étape filtration stérilisante ou répartition aseptique, dans le cas où aucun composant/excipient additionnel n’est ajouté à la substance active stabilisée.
Toutefois, les fabricants concernés bénéficiant d’un statut d’établissement pharmaceutique sur toute la chaîne de production doivent pouvoir conserver ce statut, même s’ils se conforment à la télé-déclaration d’autorisation d’activité de fabrication de substance active auprès de l’ANSM.

Partager l’article
Références
1. Extraits des Bonnes Pratiques de Fabrication, Bulletin officiel N° 2011/8 bis, publié en juillet 2011 par l’ANSM [59]
2. EudraLex – Volume 4 Good manufacturing practice (GMP) Guidelines: Part II – Basic Requirements for Active Substances used as Starting Materials; Part I – Basic Requirements for Medicinal Products;
3. Annex 1 Manufacture of Sterile Medicinal Products
4. Annex 2 Manufacture of Biological active substances and Medicinal Products for Human Use(171 KB)
Définitions
Substance active : Toute substance ou mélange de substances destinés à être utilisés pour la fabrication d’un médicament et qui, lorsqu’ils sont utilisés dans la production d’un médicament, devient un principe actif du médicament (…).
Matière première : Terme général utilisé pour désigner les matières premières de départ, les réactifs et les solvants destinés à être utilisés dans la production des intermédiaires ou des substances actives.
Produit fini : Produit dans son conditionnement final en vue de sa libération.
Equivalent : DP (Drug Product)
Produit intermédiaire : Produit partiellement manufacturé qui doit encore subir d’autres étapes de fabrication avant de devenir un produit vrac.
Produit vrac : Produit qui a subi toutes les étapes de la fabrication à l’exclusion du conditionnement final.
Equivalent : Bulk
Glossaire
BPF / GMP : Bonnes Pratiques de Fabrication / Good Manufacturing Practices
DS / API : Drug Substance / Active Pharmaceutical Ingredient ou Substance active
DP/PF : Drug Product / produit fini
AMM : Autorisation de Mise sur le Marché
ANSM : Agence Nationale de Sécurité du Médicament et des produits de santé
QP / PR : Qualified Person / Pharmacien Responsable
IMP : Investigational Medicinal Product
