


Sommaire
- Le GIC A3P Validation du Nettoyage prépare un guide pratique pour l’automne 2017 sur le Chapitre 10 de l’Annexe 15 des GMP Européennes : Présentation & discussion.
- Stratégies de regroupement des nettoyages pour les formes orales solides dans les établissements multi-produits
- La réglementation biocide appliquée aux désinfectants utilisés dans l’Industrie Pharmaceutique : ce qu’il faut savoir…
- Cahier Pratique – Comment assurer le succès d’une auto-inspection du processus d’assurance de stérilité & des procédures associées
- La cryogénie est l’étude et la production des basses températures
- Stratégie de contrôle de nettoyage dans le cadre de la fabrication de Principes Actifs pharmaceutiques en développement à usage clinique
- Le contrôle visuel indirect modernise la validation du nettoyage
- La détermination du taux de recouvrement est une étape préliminaire, nécessaire lors d’un exercice de validation du nettoyage
- Cleaning Validation for biotechnological substances : What acceptance criteria ?
- “Health-based approach” implementation for setting limits in cleaning validation for Vaccines/Biotech
Cleaning – Process validation principle
The key point before setting limits in cleaning is that cleaning is now considered as a process on its own subject to the three stage-principle developed in the FDA PV guidance (2), which are:

- Stage 1: Process Design & Development
- Stage 2 : Process Performance Qualification (PPQ)
- Stage 3 : Continued Process Verification (CPV)
Stage 1 addresses the implementation of the Health-based approach as an integral part of the design and development of the cleaning process. This Stage should describe the cleaning approach and generally includes different steps:
• Step 1: Definition of the cleaning approach
• The need for dedication or segregation of equipment,
• Matrix approaches for Products/Compounds,
• Matrix approaches for Equipment,
• Holding times definition,
• Etc.
• Step 2.1: The required level of cleanliness (Critical Cleaning Attributes)
• Visually clean and dry,
• Cleaning agent residue level,
• Maintenance agent residue level,
• Product residue level.
• Step 2.2: Design and qualification of equipment (Critical Cleaning Parameters)
• Step 3: Development of cleaning cycle(s) and associated analytical methods
New requirements – Eudralex / PICs
This article will focus on step 2.2 and the new requirements described in the Eudralex Vol.4: EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use (2015)(4):
• Chapter 5: Production (5.20)*: “A Quality Risk Management process, which includes a potency and toxicological evaluation, should be used to assess and control the cross-contamination risks presented by the products manufactured. Factors including; facility/equipment design and use, personal and material flow, microbiological controls, physico-chemical characteristics of active substance, process characteristics, cleaning processes and analytical capabilities relative to the relevant limits established from the evaluation of the products should also be taken into account. The outcome of the Quality Risk Management process should be the basis for determining the necessity for and extent to which premises and equipment should be dedicated to a particular product or product family.
This may include dedicating specific product contact parts or dedication of the entire manufacturing facility. It may be acceptable to confine manufacturing activities to a segregated, self-contained production area within a multiproduct facility, where justified.”
• Annex 15: Qualification and validation (4)
Ҥ10.6. Limits for the carryover of product residues should be based on a toxicological evaluation. The justification for the selected limits should be documented in a risk assessment which includes all the supporting references. Limits should be established for the removal of any cleaning agents used. Acceptance criteria should consider the potential cumulative effect of multiple items of equipment in the process equipment train.
• §10.6.1. Therapeutic macromolecules and peptides are known to degrade and denature when exposed to pH extremes and/or heat, and may become pharmacologically inactive.
A toxicological evaluation may therefore not be applicable in these circumstances.
• §10.6.2. If it is not feasible to test for specific product residues, other representative parameters may be selected, e.g. total organic carbon (TOC) and conductivity.
§10.14. Where a cleaning process is ineffective or is not appropriate for some equipment, dedicated equipment or other appropriate measures should be used for each product as indicated in chapters 3 and 5 of EudraLex, Volume 4, Part I”.
The new requirements include three main elements:
• The necessity to take into consideration toxicological aspects to set limits (beyond the typical historical 10 ppm or 1/1000 of therapeutic dose),
• Where necessary, the need to dedicate equipment to a specific product/solution, and
• The need to consider the cumulative effect of equipment and the associated equipment train.
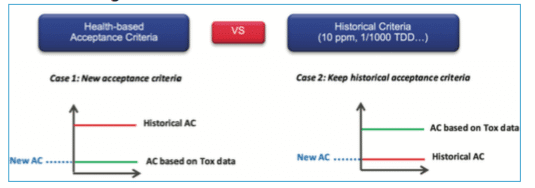
These elements put in evidence the scientific weakness of the values which are generally used in the common approaches (10 ppm, 1/1000 of therapeutic dose, or LD50). Experience shows that in general, historical values will often be more stringent than the toxicological value; however the implementation of the Health-based approach has shed light on those cases where the common approach would not result in the most stringent value. The purpose of the new regulation is not to change all historical values but to ensure that historical values are supported and confirmed by a toxicological evaluation linked to the manufactured product.
* Blue color put in evidence the necessity to have a Tox assessment, brown color the necessity to use risk management and the potential impact and green color the necessity to consider the cumulative effect.
Toxicological Evaluation
A. What must be considered?
Before launching a toxicological evaluation and invest in data collection, it’s important to define which data are necessary to set the cleaning limits. Different opinions exist; however the most complete one is to consider both APIs (and viruses, antigens…) and associated excipients.
The ingredients can be divided in three mains families:
• Products (APIs, antigens, viruses, excipients, media, adjuvants, buffers…),
• Cleaning agents,
• Maintenance agents.
B. Toxicological assessment
Once the list of ingredients has been pulled together, the toxicologist can start to assess them. This assessment is divided in four steps:
Step 1: Literature search
In this step, international literature is checked and compiled by a toxicologist. This analysis can sometimes directly lead to define if an ingredient is potentially sensitizing, mutagenic, carcinogenic, genotoxic, or has an adverse impact on reproduction. If the first assessment does not provide enough information, additional action like “readacross” or QSAR assessment are performed.
Step 2: Classification of ingredients
This step classifies the ingredients in two main families: “non-toxic” and “potentially toxic” ingredients based on information collected in step 1. Only ingredients classified in potentially toxic will follow steps 3 and 4.
Step 3: Determination of safety levels for “potentially toxic” ingredients
In this step, the toxicologist will check if existing safety limits are defined by regulation or recognized Guidelines. If these limits exist, the value used for the cleaning is derived from this pre-existing safety levels.
Step 4: PDE calculation or Safety limit definition
For this step, the toxicologist will calculate the PDE based on the most representative data (e.g., NOAEL, LOAEL, LD50) or will establish a “Safety limit” based for example on PQRI – PODP threshold, TTC…
How to use Tox data/evaluation to set cleaning limits?
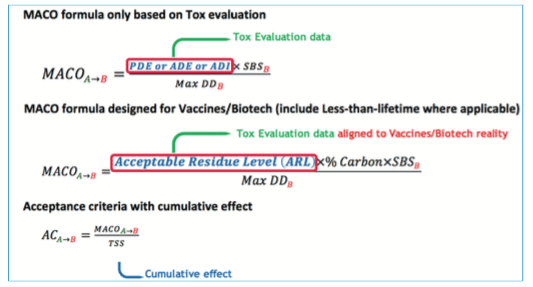
A. “MACO calculation”
At that step, a complete list of ingredients with associated PDE/Safety limits is available.
To define the final acceptance criteria, the MACO (Maximum Carry Over Allowable) must be used in conjunction with other data:
• Smallest batch size
• Maximum daily dose
• Total shared surface (also named equipment train)
B. Evolution of MACO calculation – From empiric to Tox evaluation…
This evolution can be divided in three main ways to set cleaning limits:

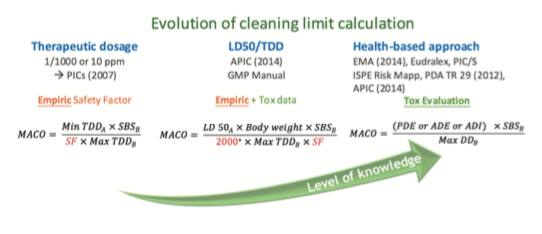
Less than Lifetime concept
This section introduces “the less than lifetime” concept for Vaccines and Biotech described in the ICH M7(6) and ICH Q3D(15). The principle is to evaluate if toxicological data that are generally defined “per day” (assuming that the ingredient is taken during the whole life everyday) can be increased to take into consideration that Vaccines injections are not made daily. As a matter of fact, in Vaccines, the worst case can be defined based on Flu Vaccines, which can be injected maximum two times per year during the whole lifetime, in comparison with other drugs that can be administered every day during lifetime.
If the toxicologist concludes that the “less than lifetime approach” can be used, then you can apply the process represented by the following schematic view. The final value used for the MACO calculation based on the toxicological evaluation and potential less than lifetime approach is named Acceptable Residue Level (ARL). For ingredients considered as “Non-Toxic”, using less than lifetime approach adds no value as the selected value (i.e. 5000 ppm) based on ICHQ3C(14) is already very high in comparison with historical approaches.
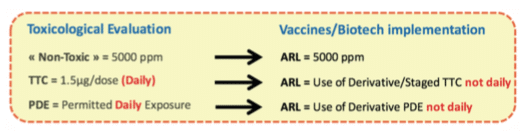
See hereunder an example of “Less than lifetime approach”:
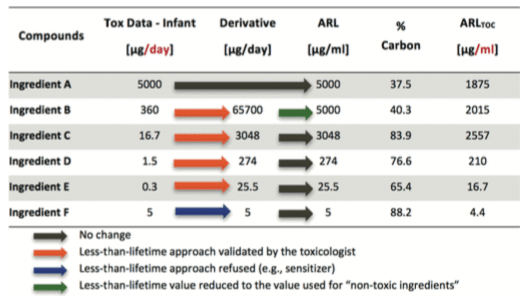
Reminder:for Biotech processes, the main tracer used is generally TOC testing; as ingredients do not content 100% organic carbon, the value must be corrected by the percentage of carbon per ingredient before setting the final value to be used in MACO calculation. If specific testing is used, this correction is not applied.
C. Equipment train – Cumulative effect
See hereunder a schematic view of an equipment train definition:
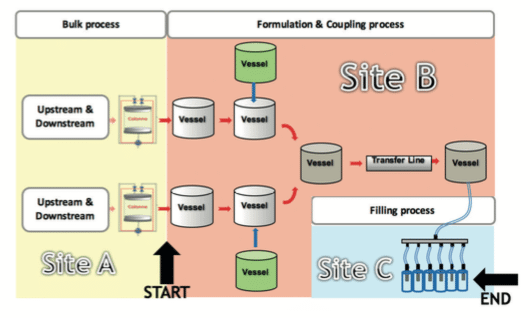
After the MACO has been aligned with the reality of the Biotech/Vaccines products, the last data required to define the acceptance criteria is the total shared surface or equipment train. As mentioned in the Eudralex Annex 15(4), it is important to consider the cumulative effect and not only the surface of the equipment alone.
The most difficult part is to define where the equipment train starts and where it finishes. To define these elements, the approach discussed in the PDA Technical Report No. 49(8) States that for the early stages of a production process (i.e. bulk manufacture) the simple use of MACO calculation with the full equipment train concept is not adequate. This is due to the fact that purification steps are present to remove different residues and to purify the product; consequently, the definition of cleaning limits may differ before and after the last purification steps.
Based on this concept, the equipment train starts right after the equipment used for the last purification step used to remove process ingredients.
As regards the equipment used before the last purification step, different approaches can be used. For example, the MACO calculation can be performed on the basis of another definition of the equipment train (i.e. TSS).
Note: sterile filtration (on 0.22 microns filter) or ultrafiltration systems that concentrate the product alone (i.e. without any other purification steps) cannot be regarded as a purification step in the context of cleaning.
On the schematic view, the starting point of the equipment train is the last column used for the purification, and the end point occurs at the stage of the needles used for the filling. The consequence of the cumulative effect is that acceptance criteria must be defined for the whole equipment train, and if this equipment train is divided between two different sites, the acceptance criteria on both sites should be the same.
Remark: another approach is to accept a higher cleaning limit on one part of the equipment train if you compensate this with stricter/lower cleaning limit on the rest of the equipment train. When doing so, the sum of the two MACO (for the two sub-equipment trains) cannot exceed the MACO calculated for the whole equipment train, and must still comply with other criteria such as visual inspection.
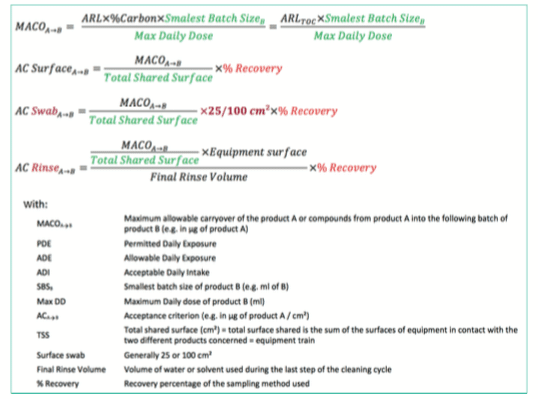
D. Summary of acceptance criteria formula based on MACO calculation
A final MACO that includes all the concepts described before can now be calculated based on following formula.
Additional information such as the swab surface (e.g., 25 or 100cm²) or the recovery percentage linked to the sampling method should be considered to set the final cleaning limits.
Finally, which limit must be used? – Toxicological or historical ones?
Toxicological evaluation can lead to very high limits and sometimes higher than the visual acceptance level. The recommendation is to perform a comparison between the historical way to calculate the acceptance criteria (i.e., 10 ppm or 1/1000 of therapeutic dose) with the new limit defined through the Health-based approach, and to use the most stringent one.

Etienne MICHEL – GSK
etienne.v.michel@gsk.com
Partager l’article
Références
(1) FDA, Guide To Inspections of Validation of Cleaning Processes (Rockville, MD, 1993).
(2) FDA, Guidance for Industry “Process Validation: General Principles and Practices”January 2011.
(3) European Commission, The Rules Governing Medicinal Products in the European Union; Volume 4 EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use (Brussels, August 2014).
(4) European Commission, Volume 4: EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use, Annex 15: Qualification and Validation (Brussels, February 2014).
(5) EMA/CHMP/CVMP/SWP/169430/2012:Guideline on Setting Health Based Exposure Limits for Use in Risk Identification in the Manufacture of Different Medicinal Products in Shared Facilities (London, November 2014).
(6) PIC/S, Recommendations on Validation Master Plan, Installation and Operational Qualification, Non-Sterile Process Validation, Cleaning Validation; PI 006-03 (September 2007).
(7) PDA, Technical Report 29 (Revised 2012), Points to Consider for Cleaning Validation, Parenteral Drug Association, Bethesda, MD, 2012.
(8) PDA, Technical Report 49, Points to Consider for Biotechnology Cleaning Validation, Parenteral Drug Association, Bethesda, MD, 2010.
(9) D.W. Layton, B.J. Mallon, D.H. Rosenblatt, M.J. Small, Deriving allowable daily intakes for systematic toxicants lacking chronic toxicity data. Reg. Tox. Pharm. 7, 96-112, 1987.
(10) G.L. Fourman & M.V. Mullen, “Determining Cleaning Validation Acceptance Limits for Pharmaceutical Manufacturing Operations,” Pharmaceutical Technology 17 (4), 54-60, 1993.
(11) Pharmtech: M. Orvais, Lai Yeo Lian, “Setting Cleaning Validation Acceptance Limits for Topical Formulations”, 2008.
(12) Active Pharmaceutical Ingredients committee (APIC), Guidance on Aspect of Cleaning Validation in Active Pharmaceutical Ingredient Plants, May 2014.
(13) International Society for Pharmaceutical Engineering (ISPE), Risk-Based Manufacture of Pharmaceutical Products, September 2010.
(14) ICH Guideline Q3C (R5) on Impurities: Guideline for residual solvents, 2011.
(15) ICH Guideline Q3D – Guideline for elemental impurities, Current step 4 version, December 2014.
(16) ICH M7 Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk – Guidance for Industry, May 2015.
