


Sommaire
- Quality Metrics: Why do manufacturers not support the FDA initiative anymore?
- Nouvelle réglementation pour dispositifs médicaux : les défis expliqués.
- Les avancées des Procédés de Hautes Pressions Hydrostatiques (HHP) dans l’obtention d’une assurance de stérilité de 10-6 : Principes & Applications pratiques
- Traitement par le peroxyde d’hydrogène vaporisé : de la décontamination à la Stérilisation ?
- Essais d’intégrité des contenants (CCIT) pour les systèmes « Single Use »
- Les nouvelles exigences des bonnes pratiques de fabrication européennes concernant la Validation du Nettoyage
- Endotoxin masking hold-time study parameter determination and performance
- Optimiser vos systèmes Qualité en changeant de paradigme : placer l’opérateur au centre des systèmes
Depuis quelques années, plusieurs changements ont été apportés aux Bonnes Pratiques de Fabrication Européennes (BPF UE) Eudralex et aux directives relatives aux produits médicinaux. Le motif de ces révisions des BPF UE par l’Agence du Médicament Européenne (EMA) est due à une revue de la “directive sur les produits médicinaux contrefaits” (Directive 2011/62/CE).

L’objectif était d’adapter les consignes des BPF UE (EMA) et de prendre en compte les nouvelles technologies de fabrication, et également d’aligner, si applicable, les BPF UE sur les autres consignes internationales (FDA, ICH).
La prévention de la contamination croisée était l’un des sujets centraux des récentes mises à jour des BPF UE. La prévention de la contamination croisée figure parmi les 10 principaux “défauts observés” de 2011 à 2013 par l’Administration sur la Réglementation de la Santé et des Médicaments (MHRA) du Royaume-Uni(1;2). Une validation solide des pratiques de nettoyage et la définition de seuils d’exposition basés sur la santé (“health based limit”) ont été identifiées comme des moyens efficaces pour prévenir la contamination croisée. En conséquence, de nouveaux ensembles d’exigences ont été ajoutés dans les mises à jour des BPF UE à propos de ces sujets.
La première partie de cet article aborde les changements clés des consignes de validation du nettoyage, y compris la définition de seuils et l’identification des résidus actifs considérés comme difficile (“worst case”) qui devraient faire partie du programme du cycle de vie du nettoyage. Enfin, la deuxième partie répond aux questions fréquemment posées par les fabricants de produits pharmaceutiques. Ces questions ont été recueillies auprès des fabricants américains et européens pendant un an et demi. Les réponses aux questions fréquemment posées s’appuient sur des références renvoyant à d’autres lectures, si nécessaires. Le présent article se concentre sur les mises à jour des BPF UE liées à l’évaluation toxicologique et pharmaceutique.
Nouvelles modifications des exigences de validation du nettoyage
Les documents suivants expliquent les nouvelles exigences en termes de validation du nettoyage :
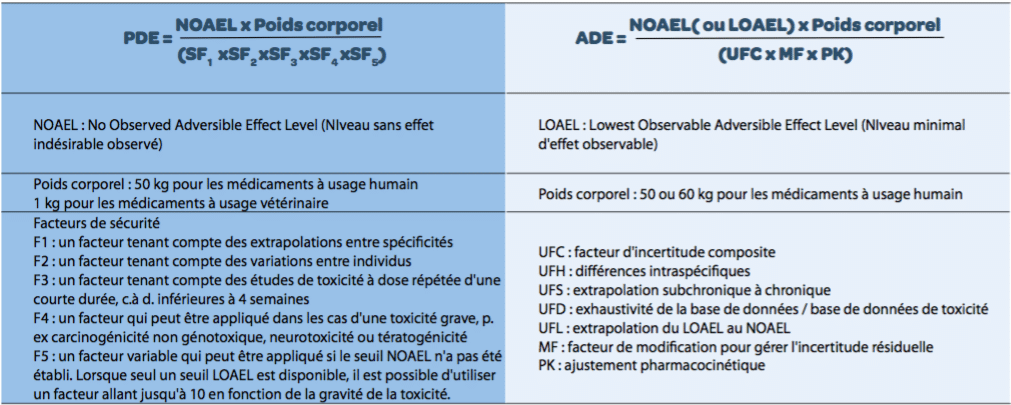
- L’EMA a révisé sa consigne “sur la définition de seuils d’exposition basés sur la santé”, applicable à compter de juin 2015(3). Les seuils limites de résidus actifs ou de détergents (seuil maximal de produit résiduel – MACO) doivent être évalués via des limites basées sur la santé utilisant l’exposition journalière admissible (PDE). La PDE représente une dose spécifique de substance qui est peu susceptible de provoquer un effet indésirable si une personne est exposée à ce niveau ou à un niveau inférieur de ladite substance chaque jour pendant toute sa vie. La PDE est calculée en fonction de l’échelle NOAEL (No Observed Adverse Effect Level, ou Niveau sans effet indésirable observé), du poids corporel et de cinq facteurs d’incertitude, parmi lesquels les données toxicologiques et pharmaceutiques cliniques ou non-cliniques. Le niveau minimal d’effet observable (LOEL) serait acceptable à utiliser si les données de NOAEL ne pouvaient pas être déterminées(4). Enfin, les données toxicologiques doivent s’inscrire dans l’évaluation de l’identification des produits enregistrés dans le site, sauf s’il est avéré que le résidu actif se dégrade et risque de devenir inactif sur le plan pharmacologique ou toxicologique.
- L’Annexe 15 des BPF UE “Quantification et Validation”, applicable à compter du 1er octobre 2015(5), impose que la validation du nettoyage soit basée sur une approche scientifique et fondée sur la gestion du risque. En conséquence, les critères de “nettoyage visuel seulement” ne sont plus acceptables. Le nombre d’exécutions de validation requises peut être déterminé via une justification de l’évaluation des risques. Pour démontrer un nettoyage robuste, des données suffisantes doivent être capturées via une surveillance continue ou une vérification périodique. La fréquence de vérification périodique sera déterminée par les résultats de l’évaluation de la qualité et du risque professionnel. Le document stipule également que le seuil de résidus actifs est calculé à l’aide de données toxicologiques (seuils basés sur la santé). À ce titre, l’efficacité de nettoyage des résidus doit être déterminés en fonction de la solubilité, de la facilité de nettoyage et de la toxicité, y compris via l’examen des données toxicologiques. Enfin, un équipement dédié devrait être envisagé lorsqu’un processus de nettoyage est inefficace pour rester sous le seuil calculé.
- Le Chapître 3 des BPF UE “Locaux et matériel” et le Chapître 5 “Production” ont également été révisés et rendus applicables à compter du 1er mars 2015. Ces documents mettent l’accent sur la prévention de la contamination croisée et sur l’évaluation toxicologique(6;7). L’organisation transitionnelle de la mise en œuvre de l’évaluation toxicologique est différente et plus stricte que celle que proposait la ligne directrice de l’EMA. En conséquence, une stratégie de mise en œuvre devrait être développée pour justifier l’éventuel manquement des échéances déterminées par les Chapîtres 3, 5 et l’Annexe 15 de Eudralex.
Impact des modifications sur la définition des seuils de résidus actifs pour les substances actives
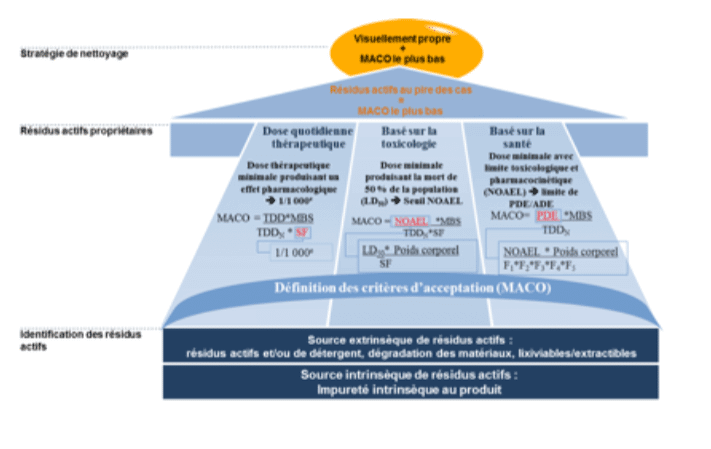
Le 1/1 000e de la dose thérapeutique minimale qui produit un effet pharmacologique (basé sur la dose) a été utilisé pour calculer le MACO(8;9). C’est ce que l’on appelle l’approche traditionnelle. Toutefois, en ce qui concerne les résidus actifs toxiques ou les composés sans données basées sur la dose, le MACO est calculé à l’aide de la dose létale 50 (LD50) orale minimale chez le rat, par exemple le détergent. D’autres études de toxicité, notamment systémique aiguë, aquatique, de cytotoxicité, etc. pourraient également être utilisées en fonction de la voie d’administration du produit et/ou de son impact sur la qualité du prochain produit fabriqué. Un facteur de sécurité basé sur les bonnes pratiques du secteur par rapport à la voie d’administration du produit et au statut du patient est également appliqué. Enfin, le seuil empirique de 10 ppm et la propreté visuelle étaient utilisés par défaut lorsque la valeur du 1/1 000e ou la valeur LD50 ne pouvaient pas être déterminées ou produisaient un résultat inutilisable.
Les nouvelles réglementations imposent que le calcul du MACO tiennent compte de l’utilisation des seuils d’exposition basés sur la santé en fonction de la méthode d’établissement de l’exposition journalière admissible (PDE) ou d’une autre méthode scientifiquement justifiée, comme décrit dans l’Annexe 3 de la directive ICH Q3C (R4) et dans l’Annexe 3 de la directive VICH GL 18(3). Il est également connu que pour une substance particulière, la valeur de PDE pourrait varier selon le toxicologue et la voie d’exposition. Un autre moyen de calculer le MACO consiste à utiliser l’exposition journalière acceptable (ADE). L’ADE est définie comme une exposition ou une dose qui est peu susceptible de provoquer un événement indésirable pour la santé si une personne doit être exposée quelle que soit la voie d’administration (p. ex. orale, dermique, contact des tissus) à une dose égale ou inférieure à ce seuil au quotidien pendant toute une vie(10). Comme pour l’ADE, la détermination d’une PDE implique l’identification des risques en passant en revue l’ensemble des données toxicologiques et pharmacologique pertinentes, la détermination du seuil NOAEL et l’utilisation de certains facteurs d’ajustement pour tenir compte de différentes incertitudes. En conséquence, l’ADE pourrait être considérée comme un équivalent de la PDE, quelle que soit la voie d’administration, en utilisant un poids corporel de 50 kg pour un adulte et un poids minimum de 1 kg pour un animal (voir Tableau 1).
Pour les résidus actifs vétérinaires, le seuil MACO pourrait être très bas pour des produits destinés à des animaux de poids légers. En conséquence, dans certains cas, la valeur de MACO pourrait être inférieure à la limite de détection ou de quantification (LOD ou LOQ). L’utilisation d’un équipement dédié semble constituer la réponse la plus simple. La définition de seuils pour tout résidu actif doit être basée sur la compréhension de la capacité de traitement de l’équipement, la méthode d’échantillonnage, la LOD de la méthode d’analyse et le seuil de résidu visuel. En conséquence, une approche justifiée basée sur le risque différenciant les produits vétérinaires, utilisés pour des animaux qui entrent dans la chaîne alimentaire humaine, pourrait éventuellement justifier une valeur de MACO plus élevée car la PDE vétérinaire devrait être inférieure à la PDE humaine. Dans le cas contraire, l’équipement devrait être dédié si et seulement si aucune justification scientifique ne peut supporter le contraire.
Les résidus actifs considérés comme pire des cas sont les substances actives présentant la plus faible valeur MACO (voir Figure 1). En conséquence, pour un équipement non dédié, si le MACO basé sur des seuils fondés sur la santé est inférieur au seuil actuellement établi en routine, une revalidation du nettoyage devrait être effectuée. Le nombre de test (“run”) à effectuer requiert une justification basée sur le risque. D’autre part, si les limites actuellement établies sont inférieures aux limites basées sur la santé, une simple justification devrait être rédigée pour démontrer que le MACO actuellement utilisé est le plus sûr pour éviter la contamination croisée. Enfin, les seuils basés sur la santé devraient généralement être supérieurs à l’approche classique ou aux seuils empiriques. Cela pourrait être utilisés par les fabricants pour justifier un phasage dans le temps pour l’identification des valeurs PDE après l’échéance de la directive.
Questions fréquemment posées par les fabricants de produits pharmaceutiques
Les récentes modifications apportées aux BPF UE ont déclenché l’examen des programmes et procédures de validation du nettoyage. Plusieurs fabricants demandent certains éclaircissements afin de comprendre comment mettre en application les dernières modifications sur leur système existant.
Voici quelques exemples de ces questions.
1. Faut-il utiliser la valeur de carbone organique total (COT) ou la conductivité pour chaque cycle de nettoyage dans la surveillance de routine ?
La collecte des données de COT ou de conductivité n’est pas requise pour chaque cycle de nettoyage. Toutefois, la fréquence de telles vérifications ou surveillances (p. ex. en utilisant le COT, la conductivité ou d’autres méthodes) devrait être liée à une évaluation basée sur le risque pour démontrer que la performance de nettoyage répond aux critères d’acceptation. D’autre part, une inspection visuelle devrait être effectuée après chaque cycle de nettoyage, dans la mesure du possible.
2. Quelle est la différence entre ADE, PDE et DJA ?
Le point de départ pour déterminer l’ADE, la PDE et la DJA est le niveau sans effet indésirable observé (NOAEL). Pour déterminer la DJA, le NOAEL est divisé par une marge de sécurité par défaut de 100. Notez qu’une exception à une marge de sécurité de 100 pourrait être acceptable dans certaines conditions(11). Les facteurs de sécurité relatifs à l’ADE, la PDE et la DJA empruntent des approches différentes. L’ADE et la PDE sont toutes deux exprimées en mg/jour pour un poids corporel de 50 kg pour un adulte et un poids minimum d’un kg pour un animal, tandis que la DJA est rapportée en mg/kg/jour. Pour une substance particulière, la valeur de PDE peut varier selon la voie d’exposition(12), tandis que l’ADE s’applique à toutes les voies d’exposition. L’ADE peut être considérée comme équivalente à la PDE dans certaines situations, mais cela doit être dûment justifié, comme expliqué ci-dessus(12).
Définition :
L’exposition journalière acceptable (ADE) est définie comme une exposition ou une dose peu susceptible de provoquer un effet indésirable sur la santé si une personne devait y être exposée par n’importe quelle voie (p. ex. orale, dermique, contact des tissus) à une dose égale ou inférieure à ce seuil quotidiennement pendant toute sa vie(13). L’exposition journalière admissible (PDE) est définie comme une dose spécifique à la substance qui est peu susceptible de provoquer un effet indésirable si une personne est exposée à une dose égale ou inférieure à ce seuil quotidiennement pendant toute sa vie(3). La dose journalière admissible (DJA) est définie comme la mesure de la quantité d’une substance particulière (initialement appliquée aux additifs alimentaires, et par la suite aux résidus d’un médicament vétérinaire ou d’un pesticide) présente dans les aliments ou dans l’eau potable qui peut être ingérée (par voie orale) quotidiennement pendant toute la vie sans risque appréciable pour la santé(14).
3. Incombe-t-il au fabricant contractuel de déterminer la valeur de PDE du produit fabriqué pour son client, par ex. pour le titulaire de l’autorisation de mise sur le marché (TAMM) ?
Les rôles et responsabilités doivent être clairement définis dans un contrat de qualité et d’approvisionnement signé entre les deux parties(15). Il incombe généralement au TAMM de déterminer la PDE des produits et/ou des agents détergents utilisés, tandis que le fabricant contractuel doit calculer le nouveau seuil maximal de produit résiduel (MACO).
Le calcul de la PDE, de l’ADE ou d’une autre valeur scientifiquement justifiée est décrit dans l’Annexe 3 du document Q3C du Conseil international de l’harmonisation des exigences techniques 4 (ICH Q3C (R4)) et dans l’Annexe 3 de la Coopération internationale sur l’harmonisation des exigences techniques applicables à l’homologation des médicaments vétérinaires 18 (VICH GL 18). Ce calcul doit être effectué par un toxicologue expérimenté qui rassemblera les données toxicologiques, pharmacologiques et cliniques ou non-cliniques afin de définir un point de départ pour calculer le NOAEL ou la dose minimale ayant un effet indésirable observé (LOAEL). Cela peut s’effectuer de deux manières différentes :
- par un examen de la fiche signalétique (SDS) et des données figurant dans la littérature concernant les matières premières entrant dans la formulation de l’ingrédient pharmaceutique actif (API) et les formulations des produits médicinaux finaux,
- via un examen des différentes données cliniques ou non-cliniques visant à évaluer si le résidu de produit ou d’agent détergent est non-toxique, toxique, sensibilisant, allergène, etc.
L’expérience a démontré que certains fabricants choisissent souvent un MACO plus strict en ajoutant un facteur de sécurité supplémentaire au calcul de la PDE en fonction de l’utilisation du produit (fréquence d’utilisation et voie d’administration). Toutefois, pour éviter de dépasser les critères d’acceptation, la procédure de nettoyage doit permettre d’atteindre des niveaux de résidus inférieurs au MACO le plus strict(16).
5. Comment peut-on calculer la PDE ou l’ADE d’un produit si la valeur de NOAEL ne peut pas être déterminée ou n’est pas disponible ?
Si la valeur de NOAEL ne peut pas être déterminée, l’utilisation de la valeur de LOAEL est acceptable(3). Si la valeur de LOAEL n’est pas disponible, il est possible de calculer une valeur de PDE ou d’ADE estimée (ou dérivée)(3). Plusieurs publications fournissent des méthodes de calcul d’une estimation de PDE pour un produit existant ou nouveau dans des installations dites “multi-produits”(17-19). Elles proposent des options permettant d’estimer la PDE ou l’ADE en fonction des seuils de préoccupation toxicologique (TTC), du niveau d’exposition professionnelle (OEL) ou de la bande d’exposition professionnelle (OEB). Notez que la PDE ou l’ADE estimées peuvent servir d’outils de hiérarchisation pour parvenir à la conformité, soit le calcul de la PDE ou l’ADE réel.
6. Si un API peut être administré par différentes voies, la PDE de l’API doit-elle être calculée pour chaque voie d’administration ?
Il incombe au toxicologue de gérer cette question et différentes réponses peuvent être acceptables selon les circonstances :
Option 1 : la PDE peut être calculée en fonction de la voie d’administration avec une biodisponibilité de 100 % (scénario dans le pire des cas). Ce type d’approche peut éventuellement aboutir à une performance de nettoyage inadéquat qui n’atteint pas le seuil MACO calculé.
Option 2 : la PDE peut être calculée en utilisant la voie d’administration qui offre la plus grande biodisponibilité. Par exemple, si un agent topique administré est fabriqué dans le même équipement qui est utilisé pour un produit administré par voie orale, la PDE des deux produits est calculée en fonction de la voie d’administration qui offre la plus grande biodisponibilité.
Option 3 : les consignes de l’Agence Européenne des Médicaments (EMA)(3) stipulent que : “La modification de la voie d’administration peut modifier la biodisponibilité ; c’est la raison pour laquelle des facteurs de correction pour l’extrapolation d’une voie à une autre devraient être appliqués si les différences sont claires (p. ex. > 40 %) en termes de biodisponibilité spécifique au mode d’administration”. En fonction de ces critères, s’il n’existe pas de différence claire en termes de biodisponibilité entre les différentes voies d’administration, il est possible de regrouper ces dernières et en conséquence, de ne pas appliquer de facteur de correction.
Cette option peut être économique pour un ingrédient pharmaceutique actif administré par différentes voies. Elle nécessite cependant une évaluation antérieure de la biodisponibilité du produit final administré par les différentes voies.
7. Est-il acceptable d’utiliser 10 ppm comme critère d’acceptation pour la limite de nettoyage si la comparaison avec l’approche de seuil basée sur la santé n’a pas été effectuée ?
La directive européenne relative à la validation du nettoyage attribue au fabricant la responsabilité du calcul du MACO en utilisant une approche basée sur la santé(3,5). Le critère de 10 ppm n’est pas conforme à l’approche scientifique préconisée par la société internationale d’ingénierie pharmaceutique pour la fabrication de produits pharmaceutiques basée sur le risque (MaPP risque de l’ISPE) ni avec la consigne de l’EMA(3,13, 20). En conséquence, l’utilisation du critère de 10 ppm sans justification adéquate par une approche basée sur la santé est inacceptable(21). En outre, les résidus au pire des cas déterminés en fonction d’une approche basée sur la santé peuvent être différents de ceux que l’on obtient en utilisant le critère des 10 ppm(20). Il est à noter que le critère des 10 ppm a été initialement utilisé dans une publication de Fourman et Mullen pour fournir une valeur par défaut et non en tant que substitut à un calcul basé sur la dose(8).
8. Est-il possible d’augmenter le seuil de nettoyage jusqu’au nouveau MACO calculé avec une approche basée sur la santé, si le nouveau MACO est supérieur au MACO actuellement utilisé sur le site ?
Si le critère de “propreté visuelle” est satisfait, la réponse est probablement “oui ” car cela augmente la “marge de sécurité” entre la capacité du processus de nettoyage et le nouveau seuil MACO. Le risque d’augmenter le seuil MACO sans justification solide peut aboutir à une observation de la part d’un auditeur, par exemple : “Pourquoi avez-vous élevé le seuil MACO si le processus de nettoyage existant fonctionnait ?” Ce type d’observation se justifie.
La réponse peut varier selon la situation :
- Si la procédure de nettoyage actuelle permet d’atteindre des niveaux de résidus inférieurs au seuil MACO actuellement utilisé sur le site, le seuil de nettoyage n’est pas augmenté jusqu’au nouveau MACO.
- Si la procédure de nettoyage actuelle et la détection par analyse sont supérieures ou égales au seuil MACO actuellement utilisé (marge de sécurité très faible ou inexistante), dans ce cas, une élévation du MACO peut être justifiée. En fait, le seuil basé sur la santé est considéré comme scientifique et s’est avéré sûr pour les patients.
9. Est-il vrai que les fabricants de biotechnologie ne devraient pas s’inquiéter du concept de PDE ?
L’Annexe 15 stipule : “10.6.1. Les macromolécules et les peptides thérapeutiques sont connus pour se dégrader et se dénaturer lorsqu’ils sont exposés à des valeurs de pH extrêmes et/ou à la chaleur, et peuvent devenir pharmacologiquement inactifs. Une évaluation toxicologique ne peut donc pas s’appliquer dans ces circonstances.” Cette phrase s’applique uniquement si le fabricant démontre que les résidus sont rendus inactifs sur le plan toxicologique ou pharmacologique par le processus de nettoyage et/ou de sterilisation en place(3,5,16).
Plusieurs fabricants de biotechnologie ont démontré, en utilisant différentes méthodes de détection (en tant qu’immunoessais spécifiques au produit tels qu’ELISA ou EIA) que les API bio-pharmaceutiques se dénaturent et se dégradent après un nettoyage, devenant inactifs sur le plan toxicologique ou pharmaceutique(22,23). D’autre part, plusieurs autres fabricants bio-pharmaceutiques déterminent la PDE de leur produit nal, notamment s’il s’agit d’un médicament conjugué (par exemple : vaccin ou anticorps conjugué) ou si le processus fait appel à des métaux lourds ou à de petites molécules(23). Remarque : si un détergent est utilisé pour le nettoyage, la PDE du détergent doit être évaluée.
Conclusion
Les récentes modifications apportées aux BPF UE ont abouti à la révision des programmes de validation et des procédures de nettoyage des fabricants. En conséquence, pour évaluer correctement l’écart par rapport aux exigences de nettoyage actuelles, il faut bien comprendre et intégrer les nouvelles exigences réglementaires aux programmes de cycle de vie du nettoyage. En outre, les régulateurs s’attendent à ce que le seuil de nettoyage utilisé par le fabricant soit justifié à l’aide d’une approche basée sur le risque afin de démontrer la sécurité pour le produit et pour le patient. Des seuils doivent être définis en fonction de la compréhension de la capacité de traitement de l’équipement, d’une méthode d’échantillonnage, d’une méthode d’analyse, d’un seuil de résidus visuels et d’un seuil de résidus pharmacologique/toxicologique.
Enfin, la validation du nettoyage et la définition de seuils de nettoyage est un processus complexe, et différentes approches (réponses) peuvent être acceptables selon les circonstances. En conséquence, les facteurs qui influencent la performance de nettoyage et définissent les limites doivent être compris afin d’éviter une procédure de nettoyage inadéquate ou un dépassement des spécifications qui aboutirait à un risque pour le patient.

Walid EL AZAB – STERIS
walid_elazab@steris.com
Partager l’article
Références
[2] Medicines & Healthcare products Regulatory Agency (MHRA) top deficiency. Accessed on April 05, 2015 at: http://webarchive.nationalarchives.gov.uk/20141205150130/http://www.mhra.gov.uk/home/groups/pl-a/documents/websiteresources/con464241.pdf [3] European Medicine Agency (EMA), Guideline on setting health based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities, November 2014
[4] Lai Yeo Lian, M. Ovais., Setting Cleaning Validation Acceptance Limits for Topical Formulations Pharmaceutical Technology, Volume 32, Issue 1, (2008)
[5] European Commission, Good Manufacturing Practice Medicinal Products for Human and Veterinary Use – Annex 15, Qualification and Validation, (2015)
[6] European Commission, Good Manufacturing Practice Medicinal Products for Human and Veterinary Use – chapter 3, Premises and equipment, (2015)
[7] European Commission, Good Manufacturing Practice Medicinal Products for Human and Veterinary Use – chapter 5, Production, (2015)
[8] Fourman, G., and Mullen, M., “Determining Cleaning Validation Acceptance Limits for Pharmaceutical Manufacturing Operations,” Pharmaceutical Technology, April 1993
[9] Active Pharmaceutical Ingredients committee (APIC), Guidance on Aspect of Cleaning Validation in Active Pharmaceutical Ingredient Plants, May 2014
[10] International Society for Pharmaceutical Engineering (ISPE), Risk-Based Manufacture of Pharmaceutical Products, September 2010
[11] Reference Dose (RfD): Description and Use in Health Risk Assessments – Background Document 1A March 15, 1993 https://www.epa.gov/iris/reference-dose-rfd-description-and-use-health-risk-assessments
[12] El Azab W., Impact of the changes to the European Good Manufacturing Practice on Cleaning Validation: Part I, GMP Journal, edition April/May, (2016)
[13] International Society for Pharmaceutical Engineering (ISPE), Risk-Based Manufacture of Pharmaceutical Products, vol.7, 1srt ed., 35-46, (2010)
[14] World Health Organization, “Principles for the safety assessment of food additives and contaminants in food,” Environmental Health Criteria, vol. 70, (1987)
[15] European Commission, Good Manufacturing Practice Medicinal Products for Human and Veterinary Use, chapter 7: Outsourced activities, (2013)
[16] Walsh A., Cleaning Validation for Biologics Can alternative approaches to the Permitted/Acceptable Daily Exposure (PDE/ADE) Be Justified?, BioPharm International Supplements, (2015). http://www.biopharminternational.com/cleaning-validation-biologicscan- alternative-approaches-permittedacceptable-daily-exposure-pdeade-be
[17] Teasdale A., D. Naumann B., Allison G., Luo W., M. Callis C., K. Shipp B., Rutter L., Seaman C., EMA Guideline on Setting Health-Based Exposure Limits, Pharmaceutical Technology, Vol. 40 (1), (2015)
[18] Dolan D.G., Naumann B.D., Sargent E.V., Maier A., Dourson M., Application of the threshold of toxicological concern concept to pharmaceutical manufacturing operations. Regul Toxicol Pharmacol, 43, 1-9, (2005)
[19] Kroes R., Renwick A.G., Cheeseman M., Kleiner J., Mangelsdorf I., Piersma A., Schilter B., Schlatter J., van Schothorst F., Vos J.G., Wurtzen G., Structure-based thresholds of toxicological concern (TTC): guidancefor application to substances present at low levels in the diet, Food and Chemical Toxicology, 42, 65–83, (2004)
[20] Crevoisier M., Lovsin Barle E., Flueckiger A., G. Dolan D., Ader A., Walsh A., Cleaning Limits—Why the 10-ppm Criterion should be Abandoned,,Pharmaceutical Technology, vol. 40 (1) (2016)
[21] Le Blanc D., Should 10 PPM be Used for Limits? (July 2016) http://cleaningvalidation.com/ les/116314751.pdf
[22] Sharnez R., Methodology for Assessing Product Inactivation During Cleaning Part I: Setting Acceptance Limits of Biopharmaceutical Product Carryover for Equipment Cleaning, Journal of Validation Technology, Vol. 18, Issue 4, (2012)
[23] Mott A., Henry B., Wyman E., Randall G., Bellorado K., Blümel M., Clark M.E., Parks M., Hayes R., Runkle S., Luo W., Methodology for Assessing Product Inactivation During Cleaning Part II: Setting Acceptance Limits of Biopharmaceutical Product Carryover for Equipment Cleaning, Journal of Validation Technology, Vol. 19, Issue 4, (2013)
