


Sommaire
- Les méthodes rapides en microbiologie. Une opportunité pour tout un chacun au sein de l’entreprise.
- Libération des lots de produits injectables : une affaire d’état !
- Cahier Pratique – Rationalisation des qualifications périodiques des autoclaves, des tunnels de dépyrogénation et des laveuses de flacons pour les formes injectables
- La détection des mycoplasmes par qPCR : les challenges de la mise en place de ce test en remplacement de la méthode réglementaire par culture décrite dans différentes pharmacopées (USP, EP, JP…)
- Environmental Monitoring Program: Hot topics in Microbiology & Best Practices.
- Quel est l’impact des désinfectants sur les contrôles d’environnement ?
- Current U.S.P. Perspectives on Microbial Identification
Depuis quelques années certains mots résonnent régulièrement dans nos oreilles dont “LEAN”, amélioration continue, innovation… Ce n’est pas toujours sans crainte qu’ils sont prononcés tant le “faire comme avant” est souvent plus “rassurant”. Il est certes plus “confortable” de ne pas trop “changer”, de prôner un certain “conservatisme”…

Pour des raisons souvent économiques, l’industrie pharmaceutique fait pourtant de gros efforts pour être dans une vraie mouvance de progrès, pour faire “aussi bien voire mieux en faisant moins ou différemment”. Ainsi quelques paradigmes ont pu être brisés. Tous ? Non, car quelques irréductibles tabous résistent encore et toujours, pas tant au progrès mais à une crainte de bouleverser le système.
Rien que d’évoquer ces tabous fait trembler les murs. C’est d’ailleurs avec crainte de froisser que cet article a été écrit. Ce n’est évidemment pas son objet… après tout, ce n’est qu’un simple article sans volonté d’imposer à quiconque la moindre consigne. Mais force est de constater que la libération des lots de produits injectables est chose sérieuse et ne peut être traitée sans déférence ni précaution. Cet article aurait été écrit ne serait-ce que dix ans auparavant, qu’il aurait certainement été voué au bucher sans aucune forme de procès… si ce n’est, à la rigueur, un procès en sorcellerie !?
La responsabilité pharmaceutique de libérer un lot ou pas est indéniablement l’une des plus grandes qui soit. Elle est bien souvent difficile. Les produits issus de procédés aseptiques ne font pas exception. Cette libération pose évidemment moins de difficultés lorsque cent pour cent des voyants sont au vert, que tous les contrôles sur le produit fini, dont le test de stérilité, sont conformes, idem pour les contrôles en cours, idem pour les qualifications, les validations… La non libération d’un lot lorsque le test de stérilité est non conforme n’est pas plus difficile. Mais que dire alors d’un lot pour lequel tous les voyants seraient au vert à l’exception d’un “tout petit” contrôle d’environnement en classe A ? La facilité serait de rebuter à grande échelle en se disant “Tuez-les tous, Dieu reconnaitra les siens”. La difficulté est de se demander ce que nous apportent les contrôles microbiologiques et ceux des particules non viables en cours de production.
Le risque déontologique serait de traiter ce sujet en liant libération des lots et économie de l’entreprise. Ce ne sera pas le cas, la ligne rouge ne sera pas franchie ! Mais au bout du compte une personne honnête, qualifiée et expérimentée devra prendre une décision en son âme et conscience à partir de “faits” mais également “d’éléments parfois moins tangibles”. L’intention de cet article est finalement de partager quelques réflexions sur le thème du contrôle d’environnement positif. Il ne donnera pas les clés de la libération ” facile” mais souhaite offrir à celui qui a la responsabilité de libérer ou pas, de prendre un peu de temps avec lui-même autour d’un sujet si difficile… mais qui ne fait absolument jamais l’objet d’aucune publication ni communication. Rappelons nous que c’est un sujet tabou !! Tout en respectant les responsabilités de chacun… mieux vaut donc en parler !
En mai 2012 a eu lieu, n’ayons pas peur de le dire, un véritable évènement. La très sérieuse Pharmacopée U.S. (U.S.P.) proposait dans sa trentecinquième édition une mise à jour à ce point ” audacieuse”, “innovante”, “inattendue”, “différente”, du chapitre <1116> (1) que si l’un de nous n’avait ne serait-ce qu’évoqué le centième, il se serait fait rapidement guillotiné ! Mais alors qui sont ces “fous furieux” qui ont travaillé pendant pas moins de sept ans sur la mise à jour de ce chapitre pour le moins avant-gardiste sur le suivi environnemental des salles propres et pourquoi ?
Ces experts du Comité Microbiologie et Assurance de la Stérilité de l’U.S.P. ont établi cette nouvelle monographie <1116> tout simplement en s’appuyant sur un constat scientifique. A partir de là, ils ne sont peut être pas si fous que ça mais tout simplement des scientifiques s’appuyant sur des faits ?
En premier lieu, ils s’étonnent de constater que les milieux gélosés recommandés pour les contrôles d’environnement sont finalement présentés comme des instruments de mesure mais à cela près qu’ils ne sont pas calibrés. Pour rappel et à titre d’exemple, la monographie <1227> (2) établit que la plupart des bactéries sont quantifiables à partir de 25 U.F.C. (Unités Formant Colonies). Que dire alors de limites recommandées à 1 ou 5 ou même à 10 U.F.C. ? Doit-on s’enorgueillir d’un prélèvement avec 4 colonies pour une limite recommandée à 5 ou trembler pour un prélèvement avec 6 colonies pour une limite toujours à 5 ? 0 colonie en classe A est-il meilleur que 2 ? Que dire également d’une limite d’alerte à 4 U.F.C. pour une limite B.P.F. (Bonnes Pratiques de Fabrication) recommandée à 5 ? La recevabilité B.P.F. d’un test n’est donc pas une fin en soi !
Certains diront qu’il s’agit bien de limites recommandées et non de spécifications imposées mais il est probable que des limites internes supérieures à celles recommandées par les B.P.F. seraient bien difficiles à défendre quand bien même elles seraient techniquement et scientifiquement plus justes.
Etant donné la capabilité des méthodes microbiologiques pasteuriennes actuelles, leur imprécision, nous ne devons pas attendre d’elles ce qu’elles ne peuvent pas offrir. Ainsi, nos experts de l’U.S.P. considèrent le suivi environnemental des microorganismes selon une approche semiquantitative et c’est là que se trouve toute l’originalité de la monographie <1116>. Avec courage, ils ont décidé d’aller jusqu’au bout de cette logique scientifique et de retirer tout simplement les limites telles que nous les connaissions jusqu’en 2012 pour les germes vivants. Oui, notre fameux comité d’experts de l’U.S.P. a retiré de la monographie <1116> les limites recommandées en U.F.C. pour préférer des prises de décisions sur la base d’informations semi-quantitatives, sur la base de taux de contamination. Dit autrement, ils proposent une lecture environnementale des Z.A.C. (Zones d’Atmosphère Contrôlée) selon leurs tendances et non plus selon une interprétation littérale d’un résultat ponctuel: une révolte ? Non, une révolution !
Attention, notre comité d’experts n’a pas perdu la tête. Il savait de toute façon qu’aucun programme environnemental ne peut prouver la stérilité de nos produits et rappelle finalement que le suivi environnemental participe indirectement à la libération d’un lot, que c’est un faisceau d’informations parmi d’autres. Il n’est pas sans ignorer que le programme environnemental n’est d’une part pas complètement fidèle, qu’il ne se suffit pas en lui-même et qu’au-delà du suivi, viennent s’accumuler un grand nombre de précautions en termes de conception et de qualification des Z.A.C. ainsi que de suivi des procédés aseptiques (simulations, filtration, encadrement). Il n’ignore pas non plus que le seul fait de prélever est en lui-même générateur de contaminations et de faux positifs.
Les amateurs de la monographie <1116>, et certainement de risques, apprécieront l’absence d’investigation jusqu’à 14 colonies: sensations fortes assurées… Il n’est pas sûr que les industriels comprennent, pas plus que les autorités européennes, ni même les américaines (3) : “En l’absence de mauvaise tendance, un seul résultat au-dessus de la limite d’action doit déclencher une évaluation et doit permettre de déterminer si des actions correctrices/préventives sont adaptées …”
Malgré l’évolution indéniable apportée par cette monographie, en toute chose il faut savoir raison garder.
La littérature sur le sujet est tout de même relativement riche avec notamment le Technical Report (T.R.) numéro 13 de la Parenteral Drug Association (4), les I.S.O. 14698 (5) sur la maîtrise de la bio contamination, la directive F.D.A. relative à la fabrication aseptique (3) et bien sûr son équivalent B.P.F. européen avec la ligne directrice numéro 1 (6). A quelques rares exceptions, dont la monographie <1116> et un peu le TR13 dans leurs dernières moutures, la très grande majorité est relativement conservatrice et recommande à ce jour des limites inférieures à la limite de quantification pour les classes A et B. Quelques éléments plus progressistes seront livrés ultérieurement.
Toujours est-il qu’au-delà de l’interprétation arithmétique des résultats environnementaux et de la pertinence des tests de stérilité, la décision du responsable pharmaceutique, en termes de contamination, sera toujours plus simple à prendre si le système qualité entourant la production aseptique est solide et pour cause ! En fait, la décision se prend elle réellement sur la base des résultats qu’il reçoit ? Ou bien les résultats qu’il reçoit ne sont qu’une information lui permettant d’apprécier si la production a eu lieu dans le périmètre connu voire “validé” (bien malin celui qui sait valider un procédé aseptique…) d’assurance de la stérilité ? Même dans un système robuste, les signaux qu’il reçoit sont relativement pauvres comparés à tous les éléments mis en oeuvre en termes d’assurance de la stérilité et l’image d’une amarre de bateau est souvent assez parlante (Cf. dessin 1).
| – Conception des Z.A.C., – Qualification des autoclaves, – Formation, – Habilitation du personnel, – Classification des Z.A.C., – Supervision du personnel, – Qualification des flux, – Validation de la filtration, – Media Fill Tests, … |  | – Suivi microbiologique environnemental, – Suivi particulaire – Test de stérilité – Dossier de lot … |
| Dessin 1 : Amarre de bateau |
Si l’amarre est bien solide, quelques cordelettes, petites ou grosses, peuvent céder, le commandant est serein et le bateau ne quittera pas le quai. Une autre image serait celle du pilote commandant un avion de ligne : un instrument peut lui indiquer une légère dérive de sa trajectoire par rapport à son plan de vol. Il peut consulter d’autres instruments, questionner des radars militaires, des radars civils, interroger d’autres avions, consulter son G.P.S. et bien entendu comparer ce qu’il voit par la vitre à ce qu’il lit sur sa carte. Si toutes les informations qu’il reçoit lui indiquent que la position de l’avion est manifestement sur la route théorique, il ignorera l’information initiale, privilégiera toutes les autres informations concordantes et gardera en tête la bonne conception de son avion, la redondance de ses systèmes de navigation… Finalement, nous n’inventons rien car les anciens avaient l’habitude de dire que les contrôles d’environnement participaient indirectement à la libération des lots, qu’ils n’étaient que le reflet de pratiques, de locaux et de compétences.
Soyons honnêtes avec nous même, personne n’est à l’aise lorsqu’une seule petite colonie est retrouvée sur les gants d’un opérateur. Patrie de Descartes oblige, nous serions tentés de prendre une décision d’ordre mathématique alors que c’est bien logiquement une décision microbiologique qui s’impose.
Les B.P.F. nous cantonnent involontairement à ce paradigme, mais cela reste un non-sens frustrant. Zéro ou une colonie, c’est pareil ! Il y a tant de raisons qu’un microorganisme ne pousse pas (Viable But Not Culturable, température, microorganisme moribond). La microbiologie n’est peut-être pas une science exacte mais c’est certainement une science quantique. A moins que tous nos produits fassent l’objet d’une stérilisation terminale ou équivalent ou d’un contrôle à cent pour cent, nos lots seront à la fois stériles et non stériles tant que nous n’apporterons pas la preuve qu’ils sont contaminés. Nos programmes environnementaux actuels, quels qu’ils soient, n’apporteront pas cette preuve. En revanche, indirectement, de manière semi-quantitative, au travers des analyses des tendances, et faute de mieux, nous indiquerons si nous sommes toujours dans notre périmètre de maîtrise ou si nous l’avons quitté.
En introduction de cette communication, il est écrit que la décision libératoire, suite à la détection d’un contrôle d’environnement est difficile mais que c’est avant tout un vrai tabou. Nos confrères nord-américains connaissent bien évidemment les mêmes difficultés mais sont “un peu” moins dans le tabou, osent “un peu” la discussion, le débat d’idées. Les communications sont rares mais infiniment plus fréquentes qu’en Europe. Ainsi, pouvons-nous lire dans la Directive Aseptique U.S. (3) : “Les niveaux d’alerte et d’action doivent être utilisés pour surveiller, maîtriser un procédé aseptique et ne doivent pas être traités comme des spécifications”.
De même, l’Aseptic Processing Working Group du très sérieux P.Q.R.I. (7) nous indique “qu’il est bien compris que les méthodes d’échantillonnage et d’incubation utilisées pour la surveillance des surfaces sont des opérations essentiellement manuelles, et du fait des interventions humaines, il peut y avoir un petit nombre de faux positifs. Pour cette raison, la détection de microorganismes sur un point critique ne doit pas nécessairement se traduire par le rejet du lot mais doit être investigué ».
Ce même Working Group du P.Q.R.I. continue en écrivant (7) “Si l’investigation ne permet pas d’expliquer un résultat positif ET qu’il n’y a pas de mauvaise tendance pour la surface critique incriminée ou avoisinante, il s’agit d’une situation clairement favorable au non rejet du lot pour cause d’un résultat positif”.
Pour être honnête, il est nécessaire de préciser que cette proximité d’idées entre les communications du P.Q.R.I. et la monographie <1116> s’explique en partie par le fait que les auteurs ou les membres étaient bien souvent les mêmes. Il est d’ailleurs probable que les travaux du P.Q.R.I. ont en fait alimenté la révolution de la monographie <1116>. Robert Friedman de la F.D.A. en va également de son couplet (8) : ” La libération des lots doit considérer 2 éléments : un seul résultat au-dessus du niveau d’action mais clairement atypique et les tendances”. Mais il tempère en rajoutant logiquement que “l’entreprise sera challengée si elle libère un lot avec un résultat O.O.S. (Out Of Specification) sur une surface et sur un opérateur, surtout s’il s’agit du même germe”.
Si nous doutons de la pertinence de nos contrôles d’environnement alors généralisons l’usage des isolateurs pourrait-on croire !
Pour peu qu’il soit convenablement conçu et exploité, un isolateur permet des activités aseptiques offrant un niveau très élevé d’assurance de la stérilité. A ce point que certaines agences évoquent ou proposent de réduire la fréquence et/ou la taille des simulations aseptiques voire d’envisager une libération paramétrique. Mais voilà, il arrive quand même que des contrôles microbiologiques d’environnement traduisent la présence d’un germe. Ce dernier est très probablement apporté lors du prélèvement lui-même ou lors des transferts ou de l’incubation. Le taux relatif de faux positifs dans les isolateurs est de fait élevé. Finalement, isolateur ou pas, investigation ou pas, les difficultés d’interprétation des résultats et de prise de décision de libération restent entières.
Si le suivi environnemental microbiologique ne tient pas toutes ses promesses, est-ce que le suivi des particules non viables peut aider dans la prise de décision que ce soit en nombre de particules ou en taille ?
Rarement oui, souvent non… mais créera certainement beaucoup de confusion ! Extrêmement rares sont les fois où contrôles des particules viables et non viables sont simultanément non conformes. Ces deux types de suivis environnementaux pèchent par des limites à la fois opposées et complémentaires : contrairement aux cultures de bactéries, de levures ou moisissures sur des milieux de cultures gélosés, les compteurs de particules sont des instruments calibrés, très sensibles mais non spécifiques.
En revanche, le suivi environnemental particulaire a un point fort de taille qui le distingue des méthodes microbiologiques pasteuriennes : son résultat est quasi instantané.
A côté de ça, le monitoring particulaire non viable présente donc l’immense défaut de ne pas être spécifique. Les intentions de ce type de suivi sont très louables sur le principe. En revanche, dans la pratique chaque démarrage de lot revient à perpétuellement ouvrir la boite de Pandore et, absence de spécificité et grande sensibilité obligent, à libérer des signaux difficilement interprétables : particules intrinsèques, pics électriques… Suite à un dépassement de limite et une faute d’investigation toujours conclusive, il est tentant de pallier cette absence de spécificité en appelant au principe de précaution et en rebutant les flacons douteux avec comme résultat : “plus je rebute et plus ce qui reste est bon”… quand on en est là…
D’une manière générale, la Ligne Directrice 1(6) est très bien informée et compréhensive: “It is accepted that it may not always be possible to demonstrate low levels of ≥5.0 μm particles at the point of fill when filling is in progress, due to the generation of particles or droplets from the product itself” et de rajouter “The occasional indication of ≥5.0 μm particle counts may be false counts due to electronic noise, stray light, coincidence, etc.”.
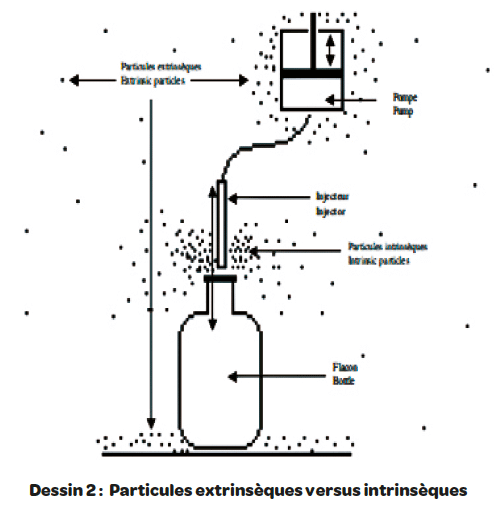
Le produit fabriqué/réparti est effectivement lui-même générateur d’un nuage de particules intrinsèques presque toujours invisibles qui par nature n’ont pas d’impact sur sa qualité (Cf. dessin 2).
Les particules extrinsèques sont quant à elles celles de l’environnement du produit. Les particules extrinsèques sont donc les seuls contaminants potentiels du produit, ce qui n’est naturellement pas le cas des particules intrinsèques. Autrement, nous serions amenés à considérer que le produit se contamine lui-même… Au-delà de cette évidence, la difficulté consiste à faire la part des choses entre les particules intrinsèques et extrinsèques lors du suivi particulaire.
En attendant des compteurs spécifiques, comment ne pas être aveuglé par les signaux “faux positifs” et faire la part des choses entre le bon grain et l’ivraie ? La Ligne Directrice 1 (6) indique “Appropriate alert and action limits should be set for the results of particulate and microbiological monitoring”. Ne s’agit-il pas là d’une invitation à l’analyse de tendance ? De plus, cette même Ligne Directrice numéro 1(6) indique également ” … consecutive or regular counting of low levels is an indicator of a possible contamination event and should be investigated”. Cette attente est certainement d’une grande portée :
• La réalité est qu’un pic isolé, même massif (surtout massif !), est pour beaucoup de Responsables Pharmaceutiques moins inquiétant qu’une succession de plus petits pics.
• La contamination particulaire a sans doute plus de sens en termes de fréquence que de taille de pic.
• La présence de pics, grands ou pas, signifie “investigation” !
• Là où l’actuelle monographie <1116> (1) est très innovante en termes de fréquence de contamination microbiologique, les autorités européennes ne le sont pas moins en termes de fréquence de contamination particulaire avec la Ligne Directrice 1 (6) mais… dès 2009!
A toutes fins utiles, il est probablement nécessaire de rappeler que non la Ligne Directrice 1 (6) n’impose pas strictement un suivi particulaire en nombre de particules. L’étude de la fréquence des pics est de fait appropriée et surtout plus pertinente en termes d’analyse d’impact et de prise de décision libératoire (9). La toute nouvelle ISO 14644-2 va d’ailleurs en ce sens avec ses approches dites alternatives. Il existe effectivement une assimilation “malheureuse” entre classification des Z.A.C. et suivi particulaire mais la Ligne Directrice 1 (6) dissipe rapidement toute confusion : “Classification should be clearly differentiated from operational process environmental monitoring”. De plus, les limites présentées au chapitre 4 sont exclusivement limitées à la classification des Z.A.C. et en aucun cas au suivi particulaire en activité. Pour s’en convaincre(6) : “Appropriate alert and action limits should be set for the results of particulate and microbiological monitoring”.
Jusque-là, l’article fait la part belle au suivi des particules dans leur composante quantitative. Bien que non spécifique, ce suivi présente tout de même une toute petite partie qualitative. Même si la littérature n’a pas cru bon à ce jour de documenter de relation entre taille et origine des particules, il est régulièrement entendu que celles de “grosses tailles”, supérieures à 5 μm, seraient d’origine intrinsèque, là où les particules de “petites tailles” seraient davantage d’origines extrinsèques.
En revanche, la littérature rappelle depuis très longtemps le lien étroit qui unit le nombre et la taille des particules non-viables avec le risque de contamination microbiologique.
Pour ce qui est du nombre, de multiples études scientifiques ont rapporté une corrélation en classe A entre le nombre de particules non viables et viables (10, 11, 12, 13, 14) et le risque associé de contamination des produits (15, 16).
Pour ce qui est de la taille, Noble, Lidwell et Kingston (17) indiquaient déjà que plus grande est la particule, plus grande est la probabilité que cette dernière porte une bactérie. Ils considèrent que les bactéries sont le plus souvent portées par des particules dont le diamètre équivalent est généralement compris entre 4 et 20 μm. Greene, Vesley, Bond et Michaelsen (18) ont spécifiquement étudié cette corrélation entre taille des particules et contamination microbienne dans les blocs opératoires. Ils ont constaté que 75,6% des contaminants microbiens sont associés avec des particules de dimensions supérieures à 5 μm.
De là à penser qu’il est possible de tolérer, sans s’interroger, des particules de 0,5 μm est bien regrettable : une augmentation de fréquence de pics, qu’il s’agisse de particules de 0,5 ou 5 μm ou autres, traduit a priori une dégradation de l’environnement, tout du moins une évolution du système de production. Au final, la taille des particules a finalement plus d’intérêt pour le producteur puisqu’elle oriente l’investigation et très peu pour le libérateur.
L’intérêt du monitoring particulaire n’est certainement pas d’évaluer la génération de particules intrinsèques, de pics électriques, de coïncidences quels que soient les tailles des particules ou des pics. En revanche, il est bien de constater précocement une contamination extrinsèque à proximité des points les plus critiques, d’alarmer le producteur sur une situation inhabituelle, de l’amener à s’interroger afin de détecter une éventuelle dégradation du système.
L’idée n’est évidemment pas de remettre en cause l’intérêt de ces contrôles en cours mais, faute de mieux, d’en rappeler quelques limites techniques, pour mieux les exploiter et au final prendre une décision de raison plus que de précaution. En attendant les progrès technologiques, l’étude des taux de contamination des particules viables et non viables en lieu et place des traditionnelles limites est une première réponse. Les progrès de la microbiologie rapide sont indéniables avec un vrai gain en sensibilité mais ce n’est pas pour autant que les compteurs de particules avec distinction des “viables et des non viables” deviendront plus spécifiques dans le proche futur. De plus, la forte proportion de faux positifs est difficile à gérer : des fibres fluorescentes par exemple peuvent être considérées comme des bactéries.
Le principe de précaution est rassurant et certainement tentant mais ce dernier n’est-il pas en fait une non décision ?
Sans cause identifiée et sur la seule base des limites B.P.F. microbiologiques ou particulaires, ce n’est pas en isolant des fractions lors de la production aseptique ou en rebutant un lot entier que les fractions restantes seront meilleures ou que les lots précédents ou suivants ne seront pas contaminés.
Les progrès des méthodes de microbiologie rapide spécifiques ET plus sensibles, et par définition plus rapides sont prometteurs. Les informations qu’elles offriront seront précieuses pour maîtriser davantage la contamination en cours de fabrication et pour accompagner la prise de décision libératoire. En attendant, seule la qualité du système fait la différence et les taux de contaminations en sont le reflet.

Olivier CHANCEL – MERIAL
olivier.chancel@merial.com
Partager l’article
Bibliographie
1 U.S.P. <1116> Microbiological Control and Monitoring of Aseptic Processing Environments.
2 U.S.P. <1227> Validation of Microbial Recovery from Pharmacopeial Articles.
3 Guidance for Industry Sterile Drug Products Produced by Aseptic Processing — Current Good Manufacturing Practice Food and Drug Administration September 2004.
4 Technical Report No. 13 (Revised) Fundamentals of an Environmental Monitoring Program, P.D.A., 2014.
5 ISO 14698 Salles propres et environnements maîtrisés apparentés 2003
6 Manufacturing Practice Medicinal Products for Human and Veterinary Use, Annex 1, Manufacture of Sterile Medicinal Products, Mars 2009.
7 Product Quality Research Institute, Aseptic Processing Working Group, Final report, 2003.
8 R. Friedman. Presentation on Sterility. Assurance Issues, P.D.A. Spring Conference, March 2002, Orlando, FL, U.S.A.
9 STP PHARMA PRATIQUES, VOLUME 18 N°6 (NOVEMBRE DÉCEMBRE 2008) – Classification et monitoring particulaire des ZAC
: interprétation de la nouvelle annexe 1 des BPF européennes, L. Bolechala, O. Chancel, J.-M. Pedebidou, L. Pisarik.
10 De Abreu, C., Pinto, T. and Oliveira, D. ‘Environmental Monitoring: A Correlation Study between Viable and Nonviable Particles in Cleanrooms,’ Journal of Pharmaceutical Science and Technology, Vol. 58, No.1, January-February 2004, pp: 45-53.
11 PDA Special Scientific Forum on Environmental Monitoring and Aseptic Processing, August 21, 2000, PDA Letter, p.1, November 2000.
12 B. Reinmüller, “Dispersion and Risk Assessment of Airborne Contaminants in Pharmaceutical Clean Rooms,” Ph.D. thesis, Bulletin No 56, Building Services Engineering, KTH, Stockholm, 2001.
13 B. Reinmüller , “Microbiological Risk Assessment of Airborne Contaminants in Clean Zones”, Bulletin No. 52, Building Services Engineering, KTH, Stockholm, 2001.
14 B. Ljungqvist and B. Reinmüller, “Hazard Analysis of Airborne Contamination in Clean Rooms—Application of a Method for Limitation of Risks,” PDA J. Pharm Sci. Technol., 49, p. 239, 1995.
15 Ljungqvist, B., and Reinmüller, B., Clean Room Design: Minimizing Contamination Through Proper Design; Interpharm Press, 1997.
16 Sinclair, C.S.. 1995. Performance of Bow/Fill/Seal Equip Under Controlled Airborne Microbial Challenges. PDA J Pharm Sci Tech. 49:294-299.
17 Noble, W. C., Lidwell, 0. M. , Kingston, D. 1963: The size distribution of airborne particles carrying microorganisms. J. Hyg., Camb. 61:385- 391.
18 Greene, V.W., Vesley, D., Bond, R.G., Michaelsen, G.S. 1962: Microbiological contamination of hospital a i r . I. Quantitative studies
Acronymes
| B.P.F. | Bonnes Pratiques de Fabrication |
| F.D.A. | Food and Drug Administration |
| I.S.O. | International Standard Organization |
| O.O.S. | Out Of Specification |
| P.Q.R.I | Product Quality Research Institute |
| T.R. | Technical Report |
| U.F.C. | Unité Formant Colonie |
| U.S. | United States |
| U.S.P. | United States Pharmacopeia |
| Z.A.C. | Zone d’Atmosphère Contrôlée |
