
Sommaire
- Le 6 Sigma et l'Excellence Opérationnelle. Juste du bon sens ?
- Combien de valeurs sont nécessaires pour avoir un échantillon représentatif ?
- Exploiter la donnée pour optimiser le pilotage d'un procédé
- Statistical modeling: The need for a reliable approach to improve process knowledge and understanding
- Bayesian approach in cosmetical research : Application to a meta-analysis on the anti-pigmenting effect of vitamin C
- Comparabilité, équivalence, similarité... Comment les statistiques peuvent nous aider à en faire la démonstration. Et bientôt la fin d'un "blind test" pour les autorités de santé et les industriels
- Le maintien du statut validé, une étape du cycle de validation
- Stratégie de validation des procédés et mise en application de l'Annexe 15 des BPF et des guidances FDA. Vérification continue des procédés (CPV)
La démarche de vérification continue des procédés ou Continued Process Verification (CPV) est un sujet qui suscite des interrogations et cela débute par les définitions. Le groupe de travail GIC A3P CPV s’est penché sur le sujet afin que cet article permette de clarifier les termes qui s’avèrent différents entre la réglementation FDA, BPF, et directives européennes, et de faire part des actions engagées par le groupe sur cette thématique.

1. Exigences et attentes réglementaires
1.1 Les textes de l’ICH
Les ICH Q8, Q9 et Q10 définissent une nouvelle approche de la qualité en production pharmaceutique. Cette approche est fondée sur une connaissance scientifique approfondie et la gestion du risque, ainsi que sur un système de gestion de la qualité approprié.
L’intégration des concepts de l’ICH dans les guides de validation de l’EMEA, et de la FDA, a entrainé une nouvelle approche en termes de validation des procédés dans l’industrie pharmaceutique.
La validation industrielle traditionnelle s’appliquait généralement à 3 lots consécutifs à échelle industrielle, puis aux lots de revalidation périodique. Les nouvelles directives européennes et américaines renforcent le fait que la validation d’un procédé n‘est pas un évènement temporel dans le développement du produit mais plutôt une validation qui dure tout le long de la vie du produit. Le cycle de vie du médicament comprend différentes étapes : la conception et le développement, la fabrication à échelle industrielle, la commercialisation jusqu’à l’arrêt de cette dernière.
Les études conduites lors du développement du procédé doivent permettre de fournir la connaissance scientifique nécessaire permettant la validation du procédé (ICH Q8 et Annexe 15 des BPF).
Les connaissances scientifiques approfondies acquises lors des différentes étapes du développement vont permettre d’approfondir la connaissance du procédé de fabrication et identifier ainsi les sources de variabilités impactant les attributs de qualité du produit fini et mieux définir ainsi la stratégie de contrôle.
ICH Q8 précise le type d’informations nécessaires dans les dossiers d’enregistrements pour démontrer la connaissance des facteurs ayant un impact sur la qualité du produit. Cette directive a également, introduit la notion du Quality by Design (QbD) et oriente ainsi vers les approches de validation des procédés fondées sur des aspects scientifiques et de management du risque. Ce dernier est bien détaillé dans l’ICH Q9.
L’ICH Q10 décrit un modèle global de système qualité pharmaceutique efficace, basé sur les concepts qualité de l’organisation internationale de normalisation (ISO) et inclut les exigences réglementaires BPF. Cette ligne directrice intervient également en complément de l’ICH Q8 “Développement pharmaceutique” et de l’ICH Q9 “Gestion du risque qualité”. L’ICH Q10 est un modèle de système qualité pharmaceutique mis en œuvre tout au long des différentes étapes du cycle de vie d’un produit. Une partie de cette directive guide en particulier dans l’amélioration continue de performance des procédés et de la qualité du produit. Cette directive met l’accent sur le maintien de l’état de control du procédé et l’amélioration continue des procédés en développant des systèmes efficaces de contrôle et de surveillance pour la performance des procédés et la qualité des produits. Cette recommandation fait partie intégrante de l’étape 3 de la validation des procédés bien détaillée dans la directive FDA. Cette approche de monitoring des procédés lors de la production de produits en phase de commercialisation intégrant l’analyse de tendance ainsi que les actions correctives et préventives (système CAPA), permet de renforcer les connaissances scientifiques du procédé acquises lors du développement.
Le respect du contenu de la ligne directrice ICH Q10 devrait faciliter l’amélioration continue et renforcer ainsi le lien entre les activités de développement pharmaceutique et de fabrication. Par conséquent, la recherche, le développement, le transfert technologique, le département de validation, et le département industriel doivent communiquer et travailler ensemble pour assurer la continuité d’informations émanant de chacune des étapes du cycle de vie du produit.
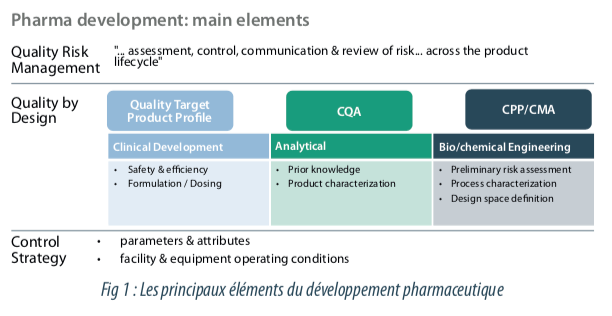
Cette étape est basée sur la vérification en continu du procédé tout au long du cycle de vie du produit pour appuyer le statut de validation du produit tel que documenté dans la revue qualité produit, en intégrant l’évaluation des tendances des procédés (Annexe 15 des BPF).
1.2 Les attentes EMEA et FDA
Dans un premier temps, une clarification des termes présentés dans les réglementations FDA, BPF et européennes est présentée ci-dessous.
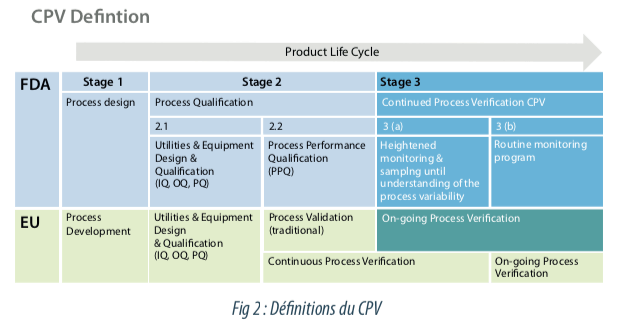
“Continous process verification” est une approche alternative pour la validation de procédé dans laquelle les performances du procédé sont monitorées et évaluées en continu lors de la fabrication. Cette évaluation continue correspond au PAT : process analytical technology (ICHQ 8).
“Continued Process Verification” correspond à l’étape 3 de la validation des procédés décrite dans les directives FDA (Guidance for Industry Process Validation: General Principles) et aussi à l’étape On Going Process Verification décrite dans les directives Européennes.
2. Considérations et problématiques rencontrées lors du déploiement de ces nouvelles directives
2.1 La démarche CPV pour les produits commercialisés (legacy product) & les nouveaux produits
La démarche CPV est applicable à tous les produits qu’ils soient des produits commercialisés ou des nouveaux produits. Néanmoins, une distinction existe. Les produits commercialisés disposent d’une validation initiale des procédés ancienne et ne disposent pas de toutes les données de développement listant les attributs qualités critiques (CQA) et les paramètres procédé critiques (PPC). Pour les produits commercialisés, il s’avère nécessaire de documenter via une analyse de risque les CQA et PPC afin de justifier la stratégie de contrôle (control strategy), qui fait partie intégrante de la démarche CPV.
Pour les nouveaux produits, le développement est en accord avec les guidelines actuelles. Ainsi, la phase 2.2 de validation traditionnelle, également appelée qualification de performance du procédé est suivie de la phase 3A de mise en place de la vérification continue du procédé sur un nombre de lots prédéfinis dans un protocole. 4 Lors de la phase 3A, les données de validation générées seront analysées statistiquement afin de confirmer la performance et la robustesse du procédé. Le statut de validation du procédé est confirmé à l’issue de la phase 3A de façon documentaire.
La phase 3B “Programme de monitoring en continue” succède à la phase 3A selon un programme et une documentation à définir. Les données suivies à fréquence périodique permettent d’identifier les dérives du procédé, d’alerter sur le risque de résultats hors spécification ce qui permet d’être proactif dans la mise en place d’actions (système CAPA). Les données collectées en phase 3B peuvent servir de données supportives à la revue périodique pour statuer sur le maintien de l’état validé.
2.2 La démarche CPV et la collecte de données, conformément aux attentes data integrity
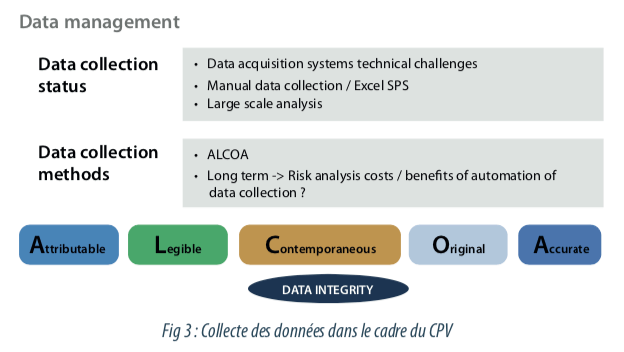
La collecte de données utilisées dans le cadre de la CPV est un véritable challenge, car elle peut être centralisée ou manuelle, et les méthodes de collectes dépendent souvent des équipements, des ateliers et des sites.
Ces données sont critiques, car elles permettent de documenter le maintien de l’état validé des procédés de fabrication. Il est donc indispensable de s’assurer du respect des règles ALCOA présentées dans la guidance “Data Integrity and Compliance With Drug CGMP, Questions and Answers, Guidance for Industry”, de la FDA de Décembre 2018. Les données complètes, cohérentes et exactes doivent être attribuables, lisibles, enregistrées en temps réel, originales ou une copie conforme et exacte.
Ces requis réglementaires peuvent être vérifiés par un processus de collecte des données documenté et validé, qu’il soit manuel ou automatisé. Une analyse de risque coût / bénéfices peut cependant être réalisée pour justifier ou non de l’implémentation d’un système de collecte automatique des données.
2.3 La démarche CPV et l’intégration dans le système qualité pharmaceutique
La mise en place de la vérification continue des procédés correspond à un profond changement de la notion de validation traditionnelle. La démarche CPV est un outil d’amélioration continue de la qualité, qui doit être intégré dans le système qualité pharmaceutique existant. Le système qualité existant peut être amené à s’adapter pour intégrer la collecte et le monitoring en continu des données.
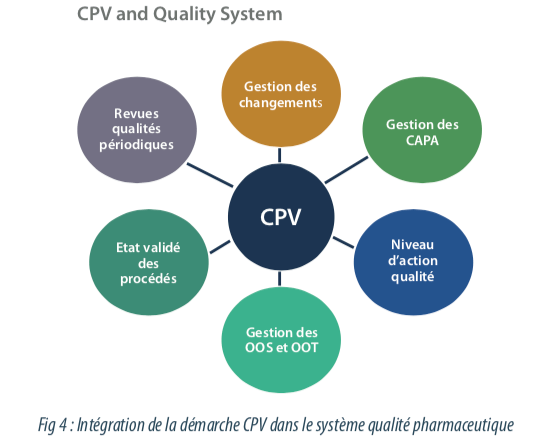
Le CPV ne remplace pas et ne se substitue pas aux processus existants. C’est un outil complémentaire pour le suivi de la maitrise de la qualité du produit, et du procédé de fabrication. Les revues annuelles produits, les processus de gestion des changements, de gestion des déviations et systèmes d’actions correctives et préventives sont des processus parallèles au CPV.
Les livrables CPV, à savoir les analyses de risque et les analyses de tendance, le plus souvent présentées dans les protocoles et rapports, peuvent être des données d’entrée, ou des Annexes des revues annuelles produit. Un processus de CPV mature peut permettre de documenter l’impact d’un ou plusieurs changements sur le procédé de fabrication. L’efficacité d’un CAPA peut également être documenté par le CPV. Le système qualité doit être adapté pour la gestion des résultats hors tendance. Deux processus peuvent coexister : le processus de gestion des résultats hors spécification, et le processus de gestion des résultats hors tendance. L’apparition d’un résultat hors tendance doit être investigué, en amont de l’apparition d’un résultat hors critère d’acceptation. C’est un outil d’amélioration continue.

 Un sondage organisé par le groupe de travail GIC A3P CPV concernant l’implémentation du CPV est actuellement en cours.
Un sondage organisé par le groupe de travail GIC A3P CPV concernant l’implémentation du CPV est actuellement en cours.
Les premières réponses indiquent que la démarche est en place pour un tiers des entreprises pharmaceutiques, en cours de déploiement pour un tiers, et en réflexion pour le dernier tiers. Pour la majorité des entreprises, la collecte des données n’est pas centralisée. Le challenge des industries restent la collecte et l’intégrité des données exploitées dans le cadre du CPV. Le CPV est un outil complémentaire pour documenter le maintien de l’état validé des procédés de fabrication, à travers la revue annuelle produit, ou une revue spécifique annuelle.
Vous pouvez d’ores et déjà répondre pour compléter cette enquête concernant le déploiement du CPV sur le site internet de l’A3P. https://a3p.org/gic-groupes-interet-commun/
Le groupe travaille également sur la rédaction d’un guide en prenant en compte les contraintes des différents types d’industries pharmaceutiques qui sera disponible prochainement.
Partager l’article


Estelle SCHUHLER – LFB
schuhlere@lfb.fr

Marinette MOREAU – VETOQUINOL
marinette.moreau@vetoquinol.com

Valérie HÉRON – SANOFI
valerie.heron@sanofi.com
Références
CPV : Continued process verification
OPV : On going process verification
PAT : Process Analytical Technology
BPF : Bonnes pratiques de fabrication
FDA: Food and Drug Administration
ICH: International Council on Harmonisation
EMEA: European Medicines Agency
CAPA: Corrective and Preventive Actions
CQA: Critical Quality Attribute
CPP / PPC: Critical Process Parameters / Paramètres Procédés Critiques
CMA : Critical Material Attribute
QI : Qualification Initiale
QO : Qualification Opérationnelle
QP : Qualification de Performance
ALCOA : Attributable, Legible, Contemporaneous, Original, Accurate / Attribuable, Lisible, Contemporaine, Originale, Exacte
OOS : Out of specification / Hors Specification
OOT : Out of Trend / Hors Tendance
Bibliographie
[ICH Q8] – International Council on Harmonisation – Pharmaceutical Development
[ICH Q9] – International Council on Harmonisation – Quality Risk Management
[ICH Q10] – International Council on Harmonisation – Pharmaceutical Quality System
EU Guidelines for GMP for Medical Products – Annex 15: Qualification & Validation; March, 2015
US FDA, Process Validation: General Principles & Practices; Jan, 2011
US FDA, Data Integrity and Compliance With Drug CGMP, Questions and Answers, Guidance for Industry», Déc. 2018
